


作者:哇塞
编辑:李宝珠
转载请联系本公众号获得授权,并标明来源
内华达大学拉斯维加斯分校的研究团队提出一种名为 ICA-Var 的多变量分析方法,该方法基于无监督机器学习流程设计,通过独立成分分析从废水数据中提取共变和时间演变的突变模式,实现了更早、更准确的变异株检测。
在过去的数年中,全球公共卫生安全面临严峻挑战。尤其自新冠疫情爆发以来,其病原体——严重急性呼吸综合征冠状病毒 2 型(SARS-CoV-2)持续进化,并相继出现多个主导疫情走势的变异株,它们具有不同的感染能力和逃避免疫应答能力,极大增加了疫情防控工作难度和医疗系统负担。
公共卫生检测和 SARS-CoV-2 基因组测序是全面检测流行变体的重要手段,但此类临床监测往往高度依赖大量实验室资源,且需个体主动参与检测,难以全面追踪 SARS-CoV-2 变异株出现和传播。尤其在医疗资源相对较少或检测意愿相对较低的地区,临床监测更容易出现检测偏差,形成防控盲区。
作为一种补充手段,利用基于废水的流行病学(Wastewater-based epidemiology, WBE)评估社区感染情况自上世纪 40 年代提出以来,已为疾病爆发预警发挥了重要作用。WBE 主要通过分析废水中人体排出的病毒痕迹,检测并追踪病毒组成和动态变化。相比临床监测,WBE 不依赖个体主动检测就能客观且无偏差地反映覆盖区域内群体感染态势,实现提前预警,具有显著的成本效益。
不过,当前主流的废水监测方法(如基于线性回归的检测方法 Freyja、COJAC 等)仍存在局限——需要基于已知变体的突变模式(如 GISAID 或 UshER 数据库中的参考序列)进行检测,若出现未在临床中表征或未被收录的新型变异株时,往往难以精准识别,这在一定程度上制约了 WBE 的检测效能。
针对于此,来自内华达大学拉斯维加斯分校的研究团队提出一种名为 ICA-Var(Independent Component Analysis of Variants) 的多变量分析方法,该方法基于无监督机器学习流程设计,通过独立成分分析( Independent Component Analysis, ICA)从废水数据中提取共变和时间演变的突变模式,实现了更早、更准确的变异株检测。
基于该方法,研究团队在 2021 年底到 2023 年间,准确检测到 Delta 变异株、Omicron 变异株和重组 XBB 变异株。此方法不仅再次印证了废水监测对疫情防控早期预警的有效性,同时也为缺乏临床监测条件下全面追踪病毒变异和传播提供了新工具。
相关研究以「Early detection of emerging SARS-CoV-2 Variants from wastewater through genome sequencing and machine learning」为题,发表于 Nature Communications。
研究亮点:
* 该方法揭示了城市和农村地区病毒变异的时空动态,证实病毒从城市向农村传播的规律,为医疗可及性差的地区或缺乏临床测序数据的情况,提供了行之有效且低成本的变异株检测范式
* 与现行金标准工具 Freyja 相比,ICA-Var 的多变量分析方法更具显著优势,在对 Delta、Omicron 及最新 EG.5、HV.1、BA.2.86 等变异株的检出时间平均提前 1-4 周

论文地址:
https://www.nature.com/articles/s41467-025-61280-5
关注公众号,后台回复「废水监测」获取完整 PDF
长周期、多点位数据采集
在本次研究中,实验所采用的废水样本来自于 2021 年 8 月至 2023 年 11 月间,从内华达州南部的城市和农村地区收集到 3,659 份废水样本。采集后的废水样本会先置于现场冰上,冷藏保存直至处理,保存时间不超过 36 小时。
在核酸提取过程中,研究团队先使用 Promega 公司生成的 Wizard Enviro Total Nucleic Kit(货号 A2991),并按照规范要求从废水样本中分离核酸。与此同时团队修改了 Promega 方案,用蛋白酶溶液裂解废水,并使用 Macherey-Nagel 公司生成的 NucleoMag Beads(货号 744970)结合游离核酸。对于总量大于 10 ng 的 RNA,研究团队使用 New England BioLabs 公司的 LunaScript RT SuperMix Kit 进行处理,用于第一链 cDNA 合成。
测序文库构建与测序方面,研究团队使用 Paragon Genomics 公司的 CleanPlex SARS-CoV-2 FLEX Panel 构建扩增子测序文库,随后文库在 Illumina NextSeq 500 或 NextSeq 1000 平台上,使用 300 循环流动槽进行测序。
测序数据的处理方面,团队先用 cutadapt 软件(4.2 版本)去除测序读段对中的 Illumina 接头序列。然后用 bwa mem 软件(0.7.17-r1188 版本)将测序读段对映射到 SARS-CoV-2 参考基因组(NC_045512.2)。接着用 fgbio TrimPrimers 工具(2.1.0 版本,硬剪切模式)去除比对读数中 Paragon Genomics 的 CleanPlex SARS-CoV-2 FLEX 扩增子引物序列。最后用 iVar variants 软件(v1.4.1 版本)检测变异位点(基于与 2020 年初始参考基因组的等位基因频率差异),并通过 samtools 软件(v1.16.1 版本)计算基因组覆盖率和读取深度。
在去除重复样本及阳性/阴性对照后,剩余 2,684 份样本用于质量控制(Quality Control, QC)分析。之后通过严格的质量控制,仅保留测序深度达到 50 倍且覆盖 SARS-CoV-2 基因组 80% 以上的废水样本用于后续分析。如下图所示:
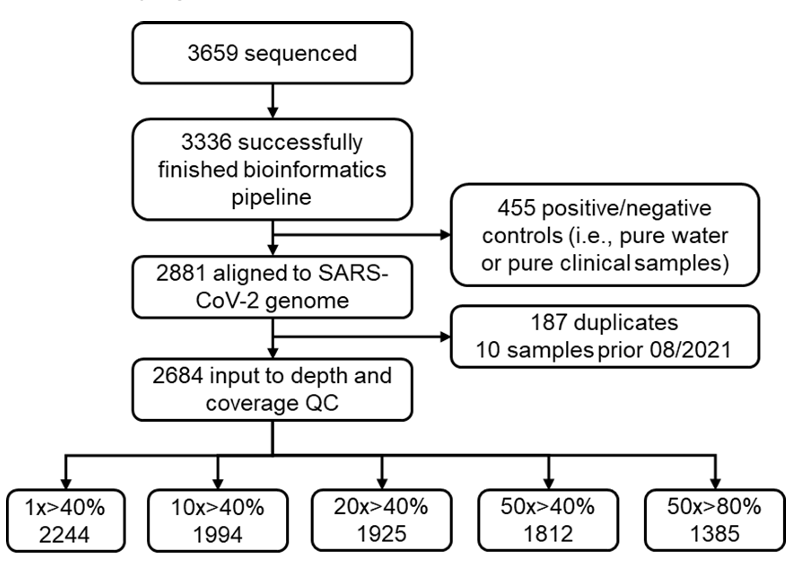
废水样本的详细筛选流程
最终,研究采用了 1,385 个高质量样本,涵盖 SARS-CoV-2 变异株的 59,422 个突变位点,用于后续分析。
为辅助验证 ICA-Var 方法的有效性,研究团队使用临床数据作为对照和参考依据,分析了从 GISAID 数据库下载的 8,810 个内华达州具有高覆盖度的临床 SARS-CoV-2 序列数据,时间覆盖 2021 年 9 月至 2023 年 11 月。
以 ICA 为核心,引入双回归方法打造新冠病毒检测新工具
ICA-Var 的核心流程,是通过独立成分分析处理废水样本中的突变频率,提取独立的共变异突变模式,然后通过双回归方法(dual-regression)将这些模式与原始样本关联,以实现对病毒变异株的追踪。如下图所示:
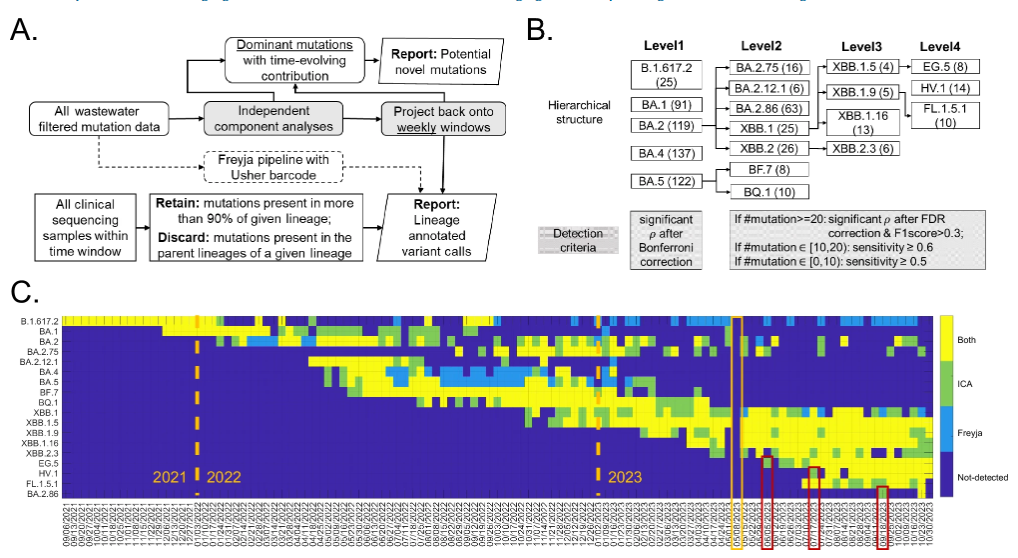
ICA-Var 方法流程,及与 Freyja 的比较
* 图中 A 为独立成分分析流程。两个矩阵分别为:每周 SARS-CoV-2 谱系检测(底行)和潜在的新突变(顶行)
* 图中 B 为 18 种关切变异株的层次结构。每种变异株的主要突变位点(即谱系定义位点)取自 covspectrum.org 总结的临床数据,括号内为主要突变的数量;阴影框为拟议流程中进行检测的标准
*图中 C 为 ICA-Var 方法与最先进工具 Freyja 的比较。对于新出现的变异株 EG.5、HV.1 和 BA.2.86,红色框表示 ICA-Var 检测时间更早;黄色框表示由于技术问题未进行废水采样的一周。
具体来说,由于废水样本中的 SARS-CoV-2 基因组信号是多种变异株混合的结果,且受到样本降解、测序误差等干扰,传统方法难以直接解析单一变异株的特征。为此,ICA-Var 的核心思路是利用独立成分分析——这一盲源分离技术,假设混合的突变信号是有多个「独立来源」线性组合而成,通过数学建模将这些独立模式从混合数据中拆解出来。
研究团队首先对数据进行了预处理,通过对废水样本的 SARS-CoV-2 基因组测序数据进行质量控制,过滤低质量读段和噪声突变,构建「突变频率矩阵」,用行代表样本,列代表突变位点,值为该位点在样本中的突变频率。之后对突变频率矩阵进行独立成分分析,将混合信号分解为独立成分,每个成分代表一组「共变异突变模式」,即某一变异株的特征性突变组合,这些突变在样本中随时间同步出现或消失。
此间,研究利用最小描述长度(Minimum Description Length, MDL)准则确定独立成分数量,利用 fastICA 算法执行独立成分分解。为确保结果可靠,他们用不同初始值重复 ICA 分析 50 次,借助 ICASSO 软件对每次运行得到的成分进行聚类和可视化,最终只保留紧密聚类对应的可靠估计作为源矩阵。
之后,为进一步确定每周的变异株情况,研究团队通过双回归方法,将独立成分分析得到的源矩阵重新投射到原始样本中,计算每个样本中个独立成分的「贡献度」,即该变异株在样本中的相对丰度,从而量化出不同变异株在时间和空间上的动态变化,如出现时间、流行趋势、城乡分布差异等。
研究团队使用全样本源矩阵作为一般线性模型(General Linear Model, GLM )中的一组源回归量,以找到与全样本源矩阵相关的每周样本的信号拆解规律。然后在第二个 GLM 中,使用每周样本的信号拆解规律作为回归量,以找到仍与全样本源矩阵相关的周特异性源矩阵。该过程生成构成对偶空间的估计值对,共同为每个周样本中的原始全样本独立成分分析源矩阵提供最佳近似。
最后,研究团队将分离出来的独立成分与临床测序数据中已知变异株进行对比并进行注释,从而成功确定其对应的变异株,或筛选出未匹配的共变突变模式,以预警新的变异株可能性。
ICA-Var 方法克服了传统方法依赖「预定义参考变异株条形码」的弊端,通过捕捉突变的共变模式,实现了比传统方法更早、更准确识别新型变异株。同时,结合了双回归分析,该方法还揭示了城乡传播差异,以及突变位点的时间进化趋势。总而言之,ICA-Var 为新冠病毒检测提供了一个更灵敏、更全面,且兼顾成本效益的工具。
检测效能超越现行金标准工具 Freyja,具备预测新变异株的潜力
为了验证和评估 ICA-Var 的性能,研究团队将其与现行的金标准工具 Freyja 进行了对比,后者是一种用于估算废水中存在的新冠病毒谱系相对丰度的工具,利用一个由界定谱系的突变所构成的「条形码」文库,来唯一确定所有已知的新冠病毒谱系,并采用深度加权、最小绝对偏差回归方法来求解谱系丰度。实验证实 ICA-Var 的多变量分析方法更具显著优势。
如下图所示。模型方法及架构部分简单阐述了 ICA-Var 能够更早地检出新变异株 EG.5、HV.1 和 BA.2.86,主要内容将在本部分将拓展来讲。
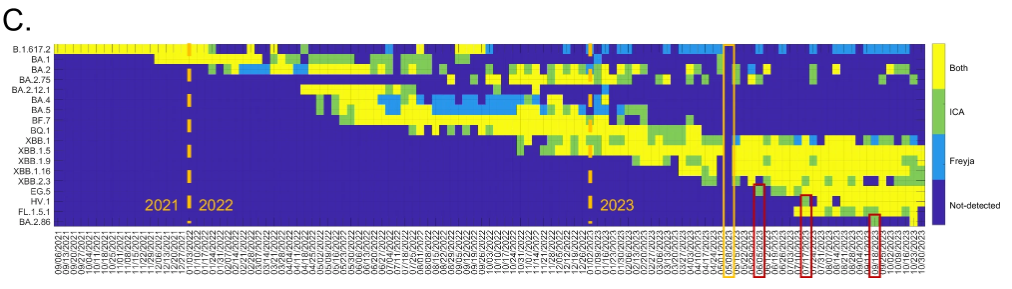
ICA-Var 与 Freyja 的比较
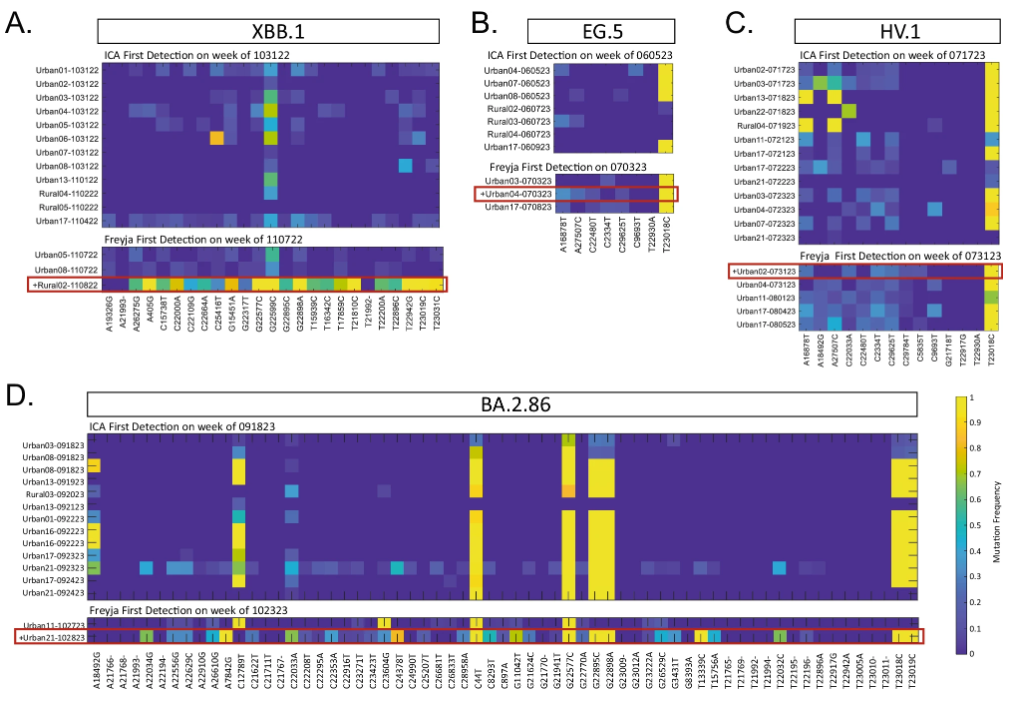
在检测 XBB.1、EG.5、HV.1 和 BA.2.86 四种变异株时 ICA-Var 与 Freyja 的对比
具体来说,在 2022 年,ICA-Var 证明能比 Freyja 早一周或数周时间,检测到 BA.2、BA.4、BA.5、BF.7、BQ.1、XBB.1 和 XBB.1.5 等变体。而在 EG.5 的检测中,ICA-Var 在 6 月 5 日那一周便检测出这种变体,但 Freyja 在 7 月 3 日才识别出 EG.5 的信号,此时废水样本丰度达 23.08%,在 8 个 EG.5 显性突变位点中已有 5 个位点显示。同样的,对于 XBB.1、HV.1 和 BA.2.86 等变异株,ICA-Var 也比 Freyja 早几周检测到。
这得益于 ICA-Var 通过整合多个样本中那些「可靠但优势突变位点流行率较低」的信息,提升了统计效力,从而实现了更早的检测。这意味着它不必依赖于单个样本中高比例的优势突变,只需汇总多个样本的微弱信号即可增强检测能力;相比之下,Freyja 则要求至少有一个单独的样本明确出现优势突变位点才能完成检测,这也意味着它更依赖于单个样本中足够明显的突变信号,对微弱或分散的信号并不敏感。
实验进一步检测了城乡样本中变异株的动态趋势。研究团队从 2022 年初开始,对内华达州南部农村地区的废水样本进行了测序和分析,并进行了全面的城乡流行病学比较,每周分别对城市和农村样本进行分析。
结果显示,ICA-Var 和 Freyja 在 18 种关切变异株中,均先在城市废水样本中检测出 16 种 SARS-CoV-2 变异株,之后才在农村样本中发现,这表明了病毒变异株通常先在城市出现,然后再扩散到农村的规律。如下图所示:
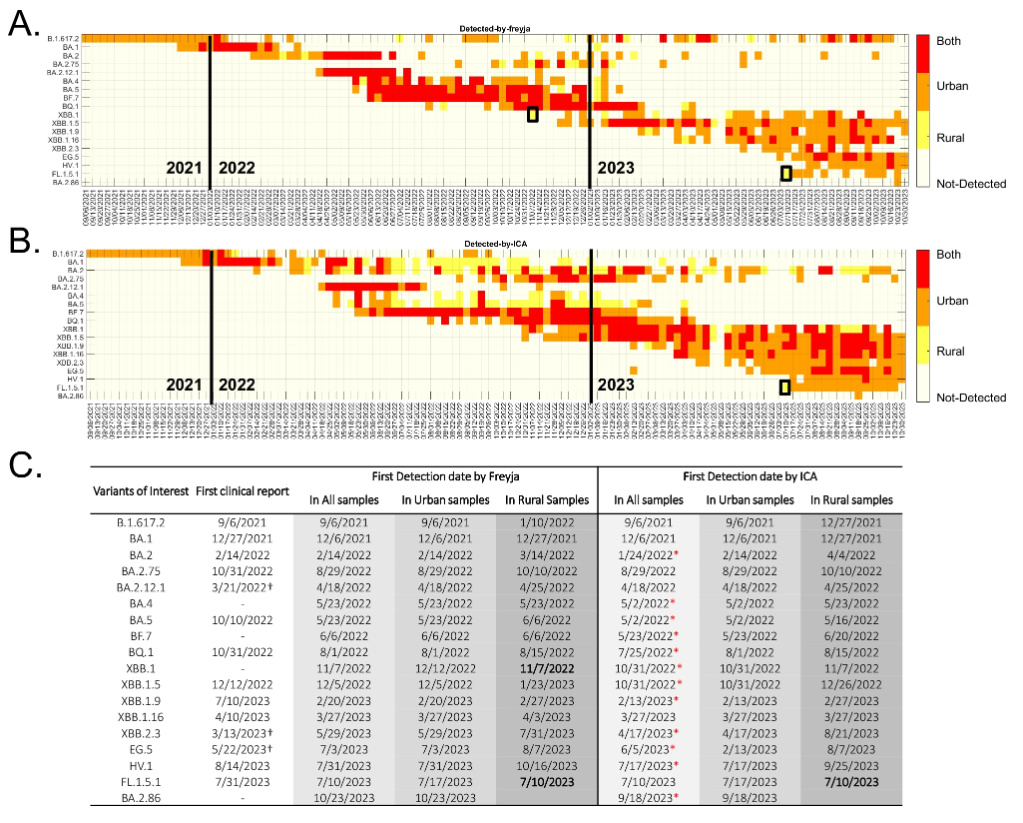
城乡废水样本的变异检测
例外的是,Freyja 最初在农村废水样本中检测到了 XBB.1,而 ICA-Var 则比之早一周在城市废水样本中发现了该变异株;两个工具都在农村废水样本中发现了 FL.1.5.1,而同期城市废水样本中,该变异株显性突变的替代等位基因频率和流行率均低得多。
研究还揭示了突变位点的时间进化趋势。研究团队将在 2021 年 8 月到 2023 年 11 月间 177 个「时间进化贡献显著的突变位点」,与 B.1.617.2、BA.1 和 XBB.1 变异体的显性突变位点进行了比较,如下图所示:
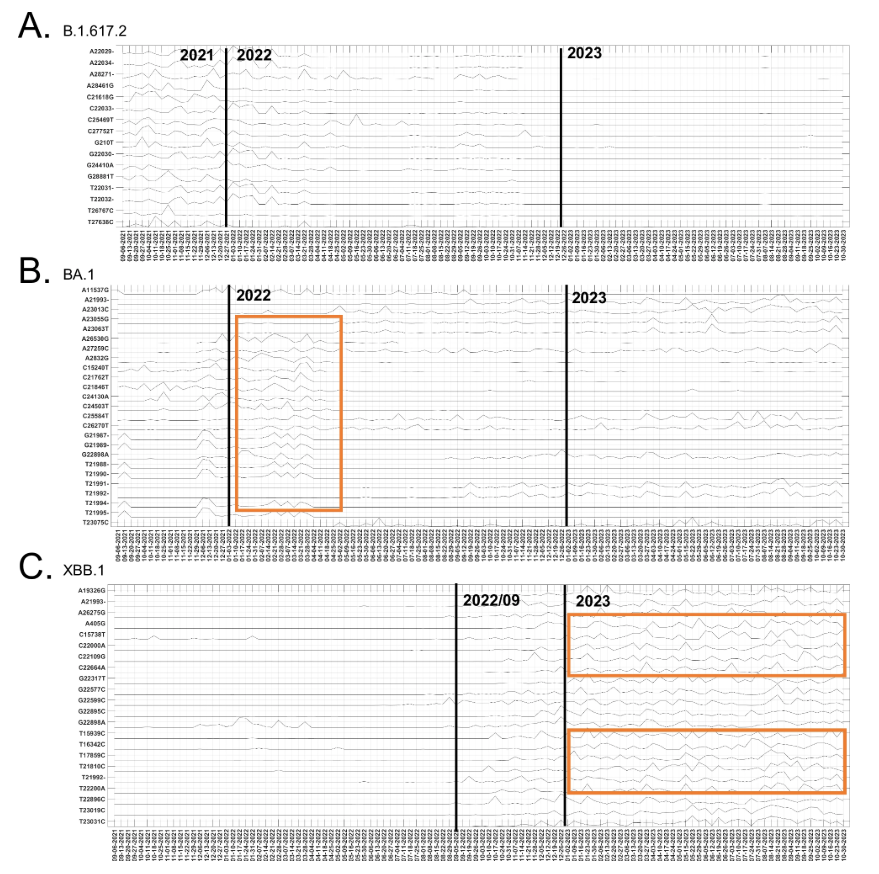
所提方法中具有显著时间进化贡献的突变
Delta 变异株(B.1.617.2)的 25 个主要突变位点中,16 个在 2021 年末出现显著波动贡献,随后在 2022 年逐渐下降;Omicron 亚型 BA.1 的相关突变贡献在 2021 年末明显增加,于 2022 年初达到峰值,部分 BA.1 突变位点的贡献在 2023 年仍有波动,且在其他 Omicron 亚谱系中被发现,如 XBB.1;XBB.1 变异株的 25 个主要突变中,22 个表现出明显的时间动态贡献,2022 年 9 月后影响显著。多个突变位点呈现相似波动模式,表明它们存在共变异,体现了 XBB.1 的重组特性。
这些分析表明了 ICA-Var 鉴定的突变位点的时间进化贡献与 Delta、Omicron 和 XBB.1 变异的临床发现是一致的,进一步说明了 ICA-Var 结果的可靠性,证明其具有识别可能导致新变体出现的新突变模式的潜力。
实验为此进行了详细验证,研究团队通过与 15 种主要变异株的显性突变位点交叉比较,筛选出 113 个潜在的新突变位点,然后采用层次聚类算法将这些突变位点分为 6 个特征簇。如下图所示:
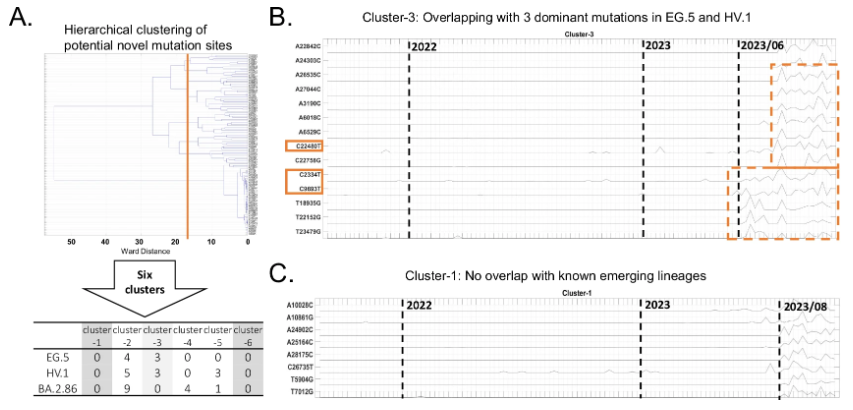
未来可能出现的突变模式
在这些特征簇中,其中 4 个(簇 2-5)的突变位点与 2023年末出现的变异株存在重叠。簇 1 和 簇 6 没有与已知突变位点的重叠突变。其中簇 1 的突变位点在 2023 年 8 月后呈现明显的共变异模式,对其中 8 个突变位点在 GISAID 的临床测序数据中验证发现,它们在临床样本中报告频率较低。因此,这些突变可能导致新型新冠病毒变异株的出现,有待临床测试进一步验证,需要密切监测。
机器学习加持,废水监测不断迭代推动高质量病毒防控
正如开篇所提,WBE 并非一种新方法,早在上世纪 40 年代,环境病毒学家就认识到通过废水中细胞培养试验获得脊髓灰质炎病毒的价值,此后 WBE 经过不断改进,进而成为用于疾病爆发早期预警的有效工具。新冠疫情爆发以来,WBE 再次为疫情防控工作起到了一定积极作用。
比如在 2023 年末,就有报道提到瑞典的研究团队通过整合污水和 COVID-19 病例的基因组检测,成功早期检测到新型 SARS-CoV-2 BA.2.86 变异株的出现。除此之外,为了更加有效、积极的利用 WBE 进行新冠病毒变体检测,不少实验室还通过开发或改进相关模型,为 WBE 提供更具成本效益的工具。
如清华大学、河北科技大学和天津生态环境监测中心的研究人员联合发表的题为「Validation of methods for enriching and detecting SRAS-CoV-2 RNA in wastewater」的研究。研究通过对比超滤(ultrafiltration)和共价亲和树脂分离(covalent affinity resin separation)两种浓缩技术,结合逆转录定量 PCR(RT-qPCR)和逆转录数字 PCR(RT-dPCR)两种检测方法,评估其在废水病毒监测中的性能。
最后,研究指出逆转录数字 PCR(RT-dPCR)方法是检测废水中低浓度 SARS-CoV-2 RNA 的更优选择,检出率更高,且对PCR抑制物具有较好耐受性。
* 论文地址:
https://link.springer.com/article/10.1007/s10311-025-01843-6
另外,加拿大阿尔伯塔大学病理和实验医学系教授李杏放教授团队发表的题为「Quantification and Differentiation of SARS-CoV-2 Variants in Wastewater for Surveillance」的研究。他们在此前为临床样本开发的 Gamma (ABG)和 Delta 多重 RT-qPCR 检测方法基础上,针对 Omicron 亚变体,利用其独特的突变,开发了 Omicron 三重 RT-qPCR 检测法,能区分五种主要的 Omicron 变异株亚系。这是单管 RT-qPCR 三重测定法在一年的时间内检测和鉴定废水样本中的所有 Omicron 亚变体的第一项研究。
* 论文地址:
https://pubs.acs.org/doi/10.1021/envhealth.3c00089
总而言之,当今世界面临严峻的公共卫生安全挑战,废水监测作为一种高效的群体监测手段,正发挥着不可替代的作用。而随着技术迭代,废水监测亦将不断发展,从早期依赖已知变异模式的靶向检测,逐步向全基因组测序、未知病原体识别等方向突破,灵敏度与覆盖面持续提升,为疫情预警、溯源及政策制定提供更精准的关键数据,成为公共卫生安全防线的重要补充。
扫码分领域获取 2023—2024 年 AI for Science 领域高质量论文汇总,内含深度解读文章 ⬇️



戳“阅读原文”,免费获取海量数据集资源!
(文:HyperAI超神经)