


作者:李宝珠
编辑:耶耶
转载请联系本公众号获得授权,并标明来源
非盈利研究机构 Arc Institute 联合 UC 伯克利、斯坦福等高校的研究团队,推出了虚拟细胞模型 STATE,能够预测干细胞、癌细胞和免疫细胞在药物、细胞因子或遗传干预下的响应情况。
众所周知,人体由不同类型的细胞组成——免疫细胞在感染发生时可引发炎症反应以抵御病原体;干细胞具有分化潜能,可生成多种组织类型;而癌细胞则通过逃避生长调控信号,实现异常增殖。尽管这些细胞在功能和形态上差异巨大,但它们几乎都拥有相同的基因组。细胞的独特性并非来自 DNA 序列本身的差异,而是源于它们如何调控和使用相同的基因信息。
换言之,细胞的特性源于基因表达的差异,而一个细胞的基因表达模式不仅决定了它属于哪种细胞类型,也决定了其所处的细胞状态,所以,通过观察基因表达的变化,便可以判断细胞是健康、发炎还是癌变。在此基础上,通过测量细胞在化学或基因干预下的转录反应,AI 模型能够学习并预测细胞在不同状态之间的转变轨迹,甚至预判未见过的干预效应。
这类「虚拟细胞」模型有望显著提升药物研发效率——在每一种药物都是一次定向干预的背景下,其能帮助科学家更精准地筛选治疗方案,引导细胞状态从疾病转向健康,同时减少副作用,从源头提升临床成功率。
如今,虚拟细胞模型成真,曾发布 Evo 系列模型的非盈利研究机构 Arc Institute 联合 UC 伯克利、斯坦福等高校的研究团队,推出了虚拟细胞模型 STATE,能够预测干细胞、癌细胞和免疫细胞在药物、细胞因子或遗传干预下的响应情况。其训练数据涵盖了来自近 1.7 亿个细胞的观察性数据,以及超过 1 亿个细胞的干预性数据,涉及 70 种不同细胞系,并整合了 Arc 虚拟细胞图谱(Virtual Cell Atlas)中的数据。实验结果显示,State 在预测干预后转录组变化的表现显著优于当前主流方法。在 Tahoe-100M 数据集的测试中,其在区分干预效果方面提升了 50%,在识别差异表达基因方面的准确率是现有模型的 2 倍。
目前,STATE 已面向非商业用途开源,相关成果以「Predicting cellular responses to perturbation across diverse contexts with State」为题发布预印本。
论文链接:https://go.hyper.ai/1UFMr
项目开源地址:https://github.com/ArcInstitute/state
融合两类数据源,涉及 70 种细胞系
STATE 由两大核心模块组成:STATE Transition(ST)和 STATE Embedding(SE),也正是基于这一多尺度框架,其能够融合两类数据源:1.67 亿个细胞的观察性数据用于训练 SE 模型,以及超过 1 亿个干预细胞数据用于训练 ST 模型。
ST 模型训练所用的单细胞干预数据集详情如下图所示,所有数据集均经过筛选,仅保留了对 19,790 个人类蛋白编码 Ensembl 基因的测量值,并统一标准化为总 UMI 深度为 10,000。

ST 模型训练所用数据集
其中:
* Tahoe-100M 数据集:大规模单细胞数据集,包含 1 亿条转录组图谱的千兆级单细胞图谱,测量了 1,100 种小分子扰动对 50 种癌症细胞系中每种细胞的影响。
Tahoe-100M 数据集下载地址:
https://go.hyper.ai/Wqbl0
* Parse-PBMC 数据集:生物科技公司 Parse Biosciences 开源的单细胞 RNA 测序(scRNA-seq)数据集,其在一次实验中对 1,152 个样本中的 1 千万个细胞进行分析,主要用于研究人类外周血单个核细胞(PBMC, Peripheral Blood Mononuclear Cells) 在不同条件下的基因表达特征。
Parse-PBMC 数据集下载地址:
https://go.hyper.ai/20nBg
SE 模型是在 1.67 亿个人类细胞上进行训练的,数据来源如下图所示,为了避免在语境泛化基准测试中发生数据泄漏,研究人员在训练中仅使用了 Tahoe 数据集中的 20 个细胞系,并将另外 5 个细胞系作为保留测试集

SE 模型训练所用数据集
其中,Arc Institute 不久前发布的大规模人类单细胞表达数据集 scBaseCount,包含超过 4 千万个人类细胞,覆盖多个器官、细胞系和病理状态。本次研究中,在处理 scBaseCount 数据时,研究人员筛选了每个细胞中具有至少 1,000 个非零表达值和 2,000 个 UMI 的细胞。
基于 Transformer 构建的多尺度框架 STATE
STATE 能够预测细胞受到扰动后的下游转录组响应,包括基因表达的变化、差异表达基因,以及整体扰动效应的强度。该架构整合了多个层次的信息:
* 分子层面:使用嵌入表示不同实验和物种中各个基因的特征;
* 细胞层面:使用嵌入表示单个细胞的转录组状态,既可以是细胞的 log-normalized 表达谱,也可以是由 STATE Embedding(SE)模型生成的嵌入;
* 群体层面:STATE Transition(ST)模型在细胞集合上学习扰动效应。
其中,ST 基于 Transformer 架构,通过自注意力机制建模干预在细胞集合中的转化过程,每个细胞可用原始基因表达或嵌入向量表示。SE 模块则在多种异质性数据集上预训练而成,能够学习细胞间的表达差异,生成对技术噪音鲁棒、同时对干预响应高度敏感的表达性向量。借助自注意力机制,ST 模型无需显式分布假设,便能灵活捕捉复杂的生物变异性。
如下图所示,作为一个多尺度机器学习框架,STATE 可以在基因、单个细胞和细胞群体等多个层面上运行。其中,ST 模型通过在共享协变量(shared covariates,如扰动类型、细胞环境和 batch)下分组的扰动与未扰动细胞群体集合上进行训练,来学习扰动效应。ST 模型可以直接处理基因表达谱,也可以处理来自 SE 模型的紧凑型细胞表示,SE 模型从大规模观察数据中学习富含信息的嵌入表示。
同时,该多尺度架构使 ST 能够有效地在计算机中模拟 Perturb-seq 实验,并支持后续分析任务,如表达量估计、差异表达分析和扰动效应量估计。
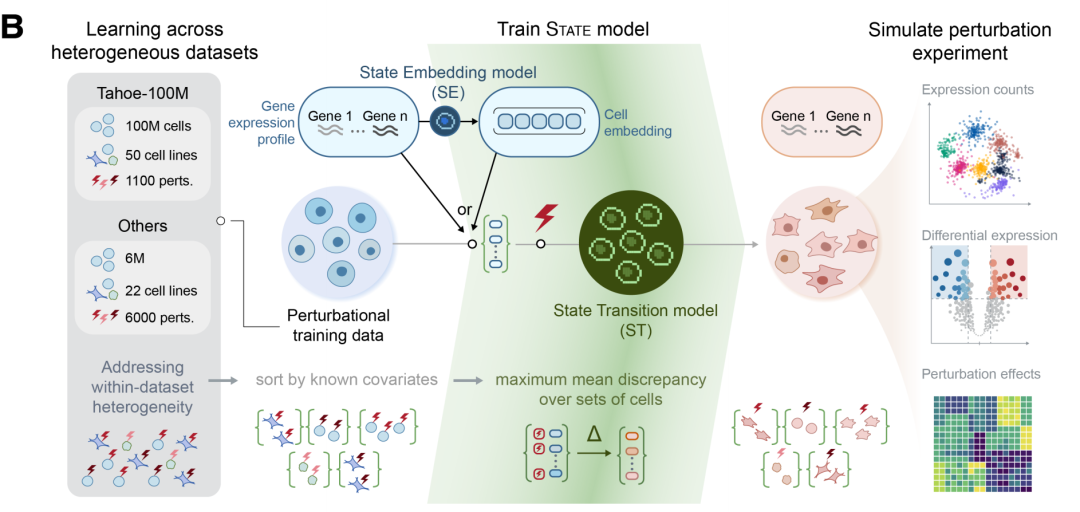
STATE 基本框架
ST 模型框架如下图所示,其输入为未扰动细胞群体的集合(unperturbed cell populations)和扰动标签(perturbation labels),输出为对应的扰动后细胞群体(perturbed cell populations)。当细胞由基因表达谱表示时,ST 可以直接预测单细胞层面的转录组;当使用 STATE 嵌入作为输入时,ST 首先预测输出嵌入,然后通过一个多层感知机(MLP)将其解码为转录组。
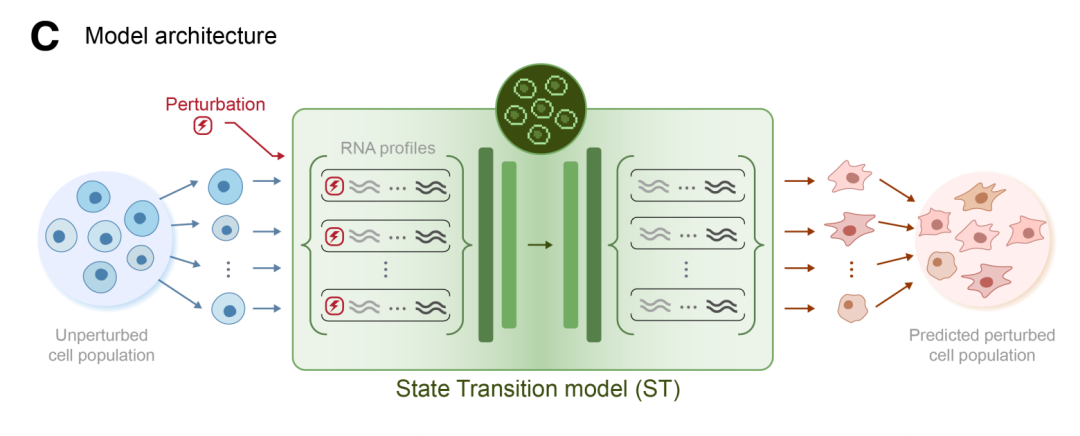
ST 模型框架
ST 模型的训练目标是最小化预测的扰动细胞转录组与真实观测数据之间的最大均值差异(MMD)损失。尽管 ST 是在细胞分布层面上学习扰动效应,但其仍然为每一个具体细胞预测扰动后的表达谱。这一特性对于捕捉扰动群体中细胞的分布结构至关重要。
实验证明,在不超过某一阈值的范围内,增加细胞集合的规模能显著降低验证损失,效果明显优于对单个细胞进行建模。此外,移除自注意力机制会导致性能下降,如下图 D 所示,这进一步说明了基于集合的灵活自注意机制在建模扰动响应中的细胞异质性方面具有重要价值。
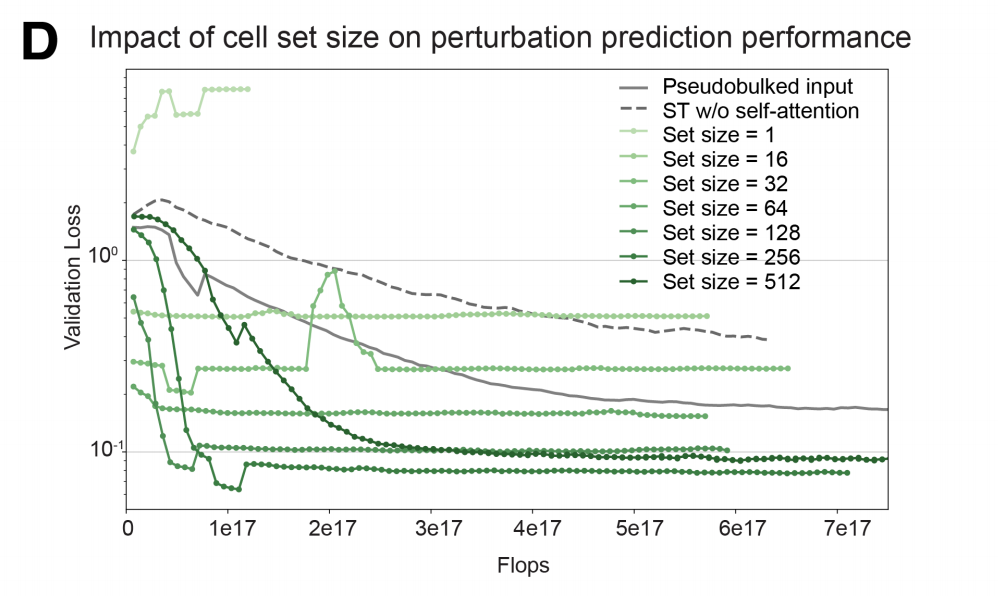
细胞集合规模对扰动预测性能的影响
SE 模型是对 ST 模型的补充,旨在学习细胞嵌入,优化捕捉细胞类型特异性的基因表达模式,如下图 A 所示。SE 在数据量较少或实验噪声较大的情况下尤为有用。与 ST 结合使用时,SE 提供了一个更加平滑的细胞状态空间,这一嵌入是基于大量观察性单细胞数据库学习而成的,相当于是间接利用丰富的观察性单细胞数据来提升扰动响应预测的准确性,特别是在干预数据有限的情况下效果显著。
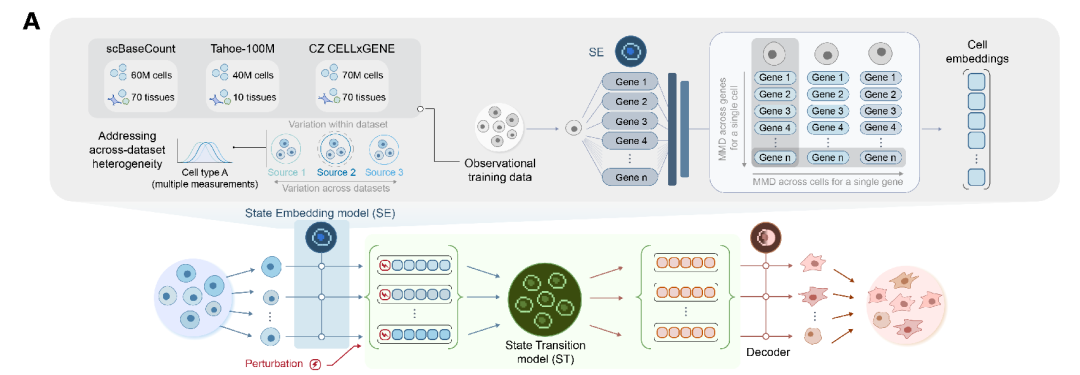
SE 模型架构
在架构上,SE 编码器是一个密集的双向 Transformer,训练目标是预测对数归一化的基因表达。SE 解码器是一个较小且专门设计的多层感知机(MLP),基于学习到的细胞嵌入和目标基因嵌入的组合来预测基因表达,这种架构上的不对称设计,使得模型学习到具有生物学基础且具备良好泛化能力的细胞状态。
STATE 在跨细胞环境的扰动效应预测上全面领先
研究人员将 STATE 与多种 baseline 模型进行了对比评测,其中包括 3 种机器学习模型:CPA、scVI 和 scGPT,并在在化学、信号传导和基因扰动数据集上进行了评估。其评估框架覆盖了 Perturb-Seq 实验的 3 个核心输出类别:基因表达计数(gene expression counts),差异表达统计信息(differential expression statistics),扰动效应的整体强度(magnitude of the perturbation effect)。
为全面评估模型在这些维度上的表现,研究人员开发了一套评估指标体系 Cell-Eval,如下图 C 所示,这些指标既具表达性,也具有生物学解释力,能够提供互补的评估视角。例如 DEGs 的重叠程度有助于将预测结果与特定通路联系起来,赋予其生物学意义;而扰动判别分数能更敏感地捕捉扰动效应的细粒度变化,反映预测结果与真实扰动效应之间的相似度。
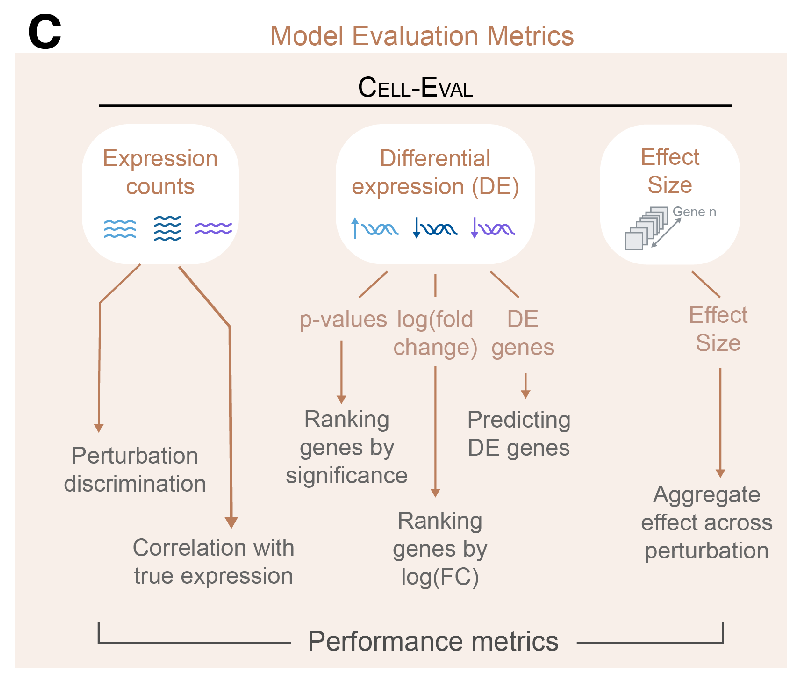
虚拟细胞建模评估框架 Cell-Eval
具体评估中,针对扰动实验,模型必须能够有效地区分不同扰动的效应。对此,研究人员使用了一种改编自 Wu 等人于 2024 年提出的扰动判别分数评估方法,该方法通过比较预测的扰动后表达谱与真实扰动结果的相似性,对扰动效果进行排序。结果显示,STATE 模型在 Tahoe 和 PBMC 数据集上的表现分别提升了 54% 和 29%,如下图 D 所示。
为直接评估对基因表达计数预测的准确性,研究人员计算了观测到的扰动引起的表达变化与模型预测值之间的 Pearson 相关系数。在这一指标上,STATE 模型在 Tahoe 数据集上的表现比基线模型高出 63%,在 PBMC 数据集上高出 47%,如下图 E 所示。
为评估模型预测的差异表达(DE)基因的 p 值,研究人员首先使用实验中观测到的扰动数据计算出真实的显著差异表达基因,并设定 FDR 阈值为 0.05。随后将模型预测所生成的 p 值与真实的显著性水平进行比较,并绘制精确率-召回率(precision-recall, PR)曲线。通过计算 PR 曲线下面积(AUPRC)可发现 STATE 在所有数据集上始终优于所有基线模型,如下图 F 所示。
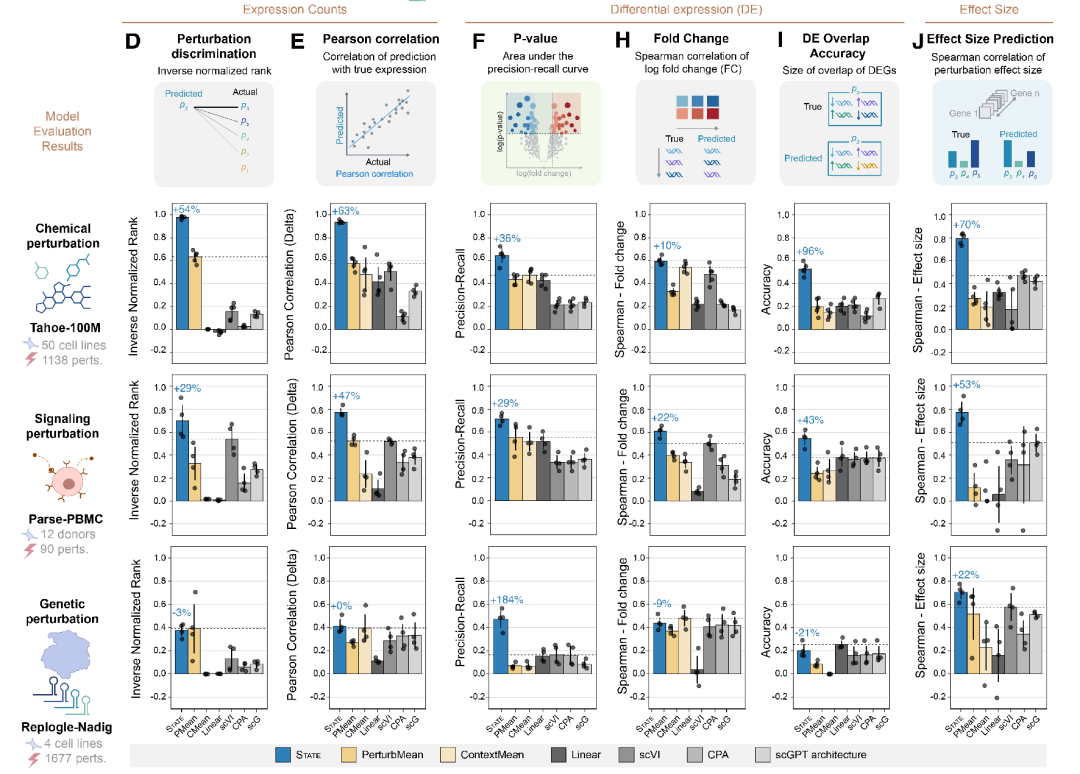
STATE 与基线模型在多个评测任务上的性能对比
STATE 模型在基因扰动数据集上的 AUPRC(精确率-召回率曲线下面积)比排名第二的模型高出 184%,这一结果在各模型于不同数据集上的 PR 曲线中表现得非常明显,如下图 G 所示。
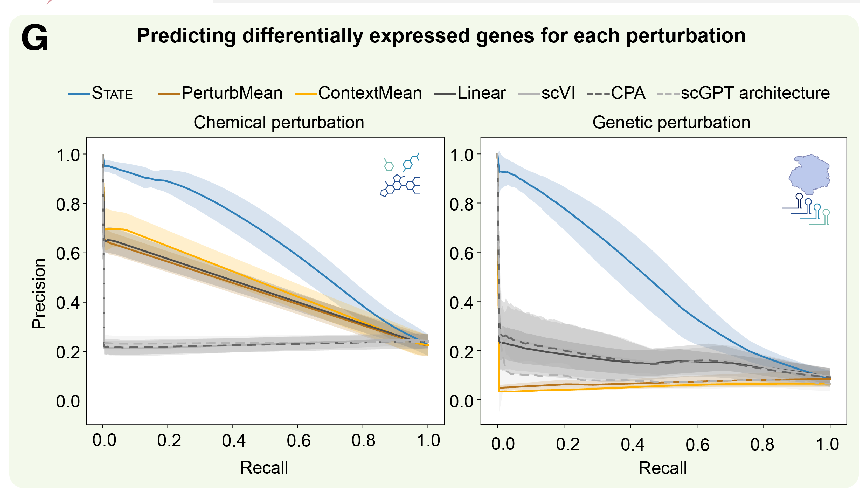
预测每种扰动下差异表达的基因
另外值得一提的是,STATE 还支持零样本预测(zero-shot),即在模型训练时未见过扰动数据的新细胞环境中,也能准确预测扰动效应,如下图所示。
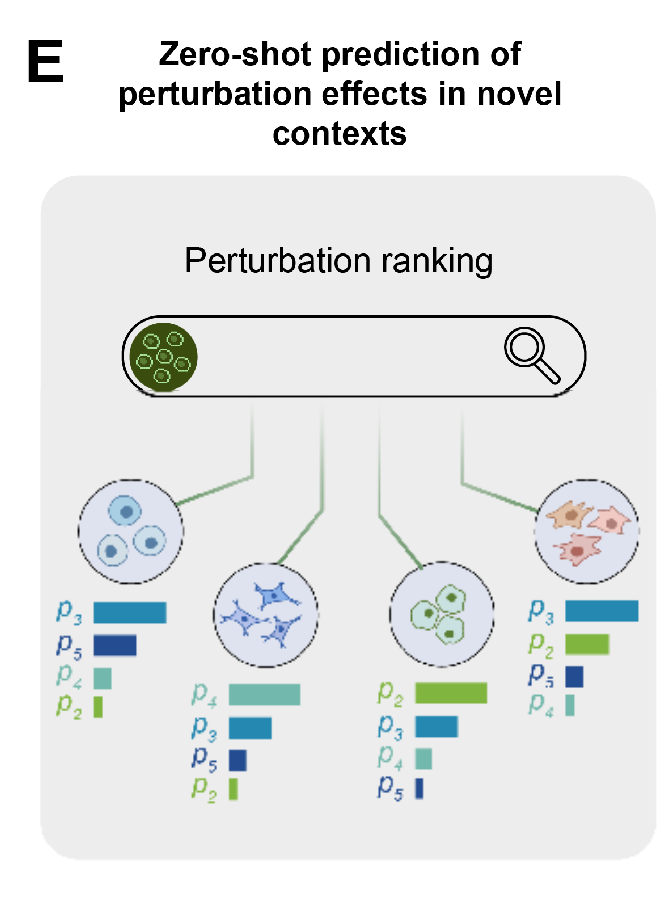
STATE 可实现零样本预测
更进一步地,为展示 STATE 的实际应用场景,研究人员评估了其检测细胞类型特异性差异表达的能力,聚焦于 Tahoe-100M 数据集中 5 个细胞系如下图 A 所示。
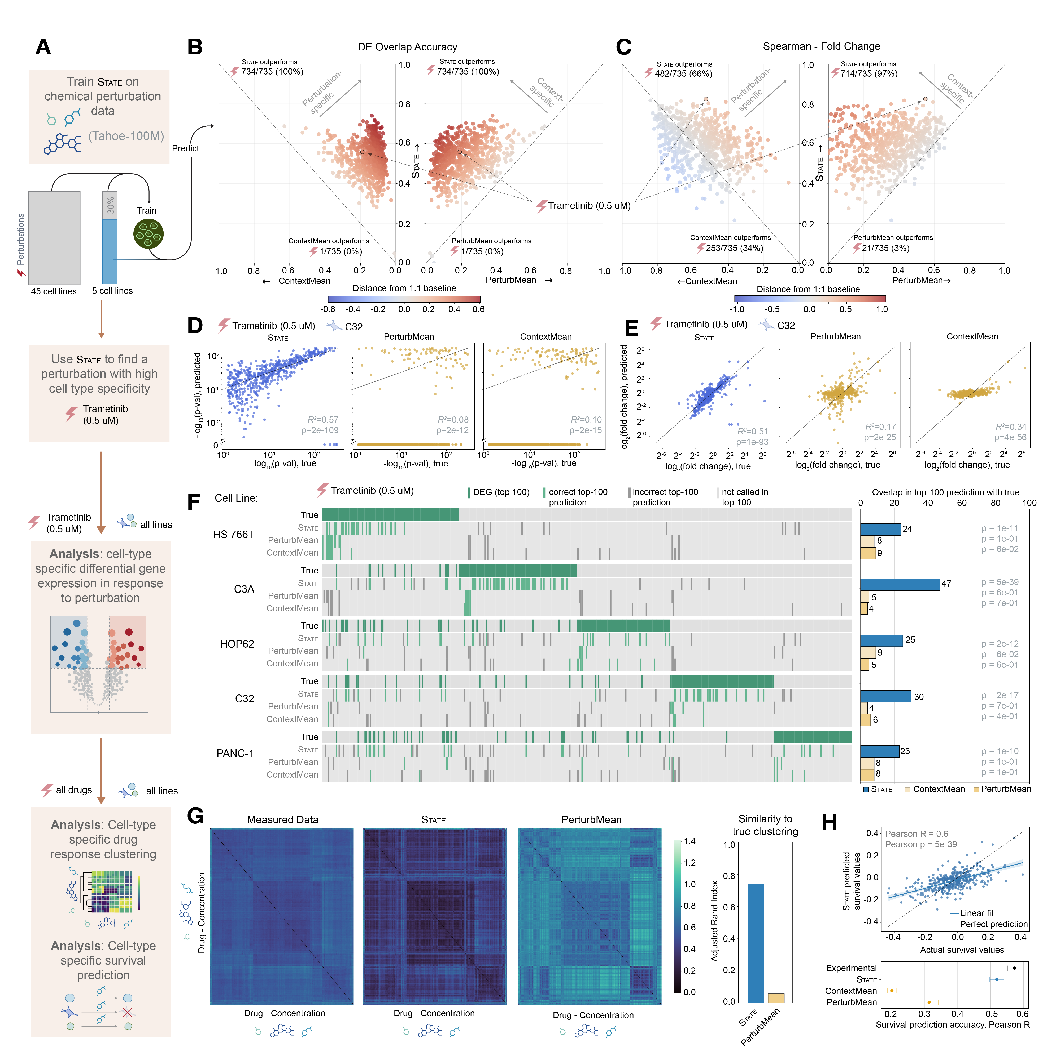
State 能检测扰动引起的细胞类型特异性基因表达变化
研究人员通过对比 STATE 与两个基线模型的预测结果在差异表达基因的重叠程度以及 log fold change 的 Spearman 相关系数,来识别出具有强细胞类型特异性的扰动条件。若性能优于「扰动均值」基线,说明 STATE 学会了特定于某一细胞类型的扰动效应;若优于「环境均值」基线,则表明模型能够区分同一细胞系中不同扰动的影响,而不是简单地预测每个细胞系的平均表达水平。
在所有扰动条件中,STATE 始终表现出更强的能力,能更准确地还原差异表达基因的 log fold change 的真实排序,显著优于环境均值和扰动均值两种基线模型,如上图 B 所示。
总结来看,研究团队提出,STATE 是首个在细胞环境泛化任务中,几乎在所有指标和多个数据集上均超越简单基线(如均值模型或线性模型)的机器学习模型。此外,结合了细胞嵌入模型 SE 生成的嵌入,使得在新细胞环境中实现更有效的零样本扰动效应预测成为可能。
非营利性研究机构 Arc Institute 发布一系列重磅成果
由知名移动支付公司 Stripe 联合创始人兼 CEO Patrick Collison 和斯坦福大学生物化学助理教授 Silvana Konermann 夫妻二人,联合加州大学伯克利分校生物工程助理教授徐安祺(Patrick D. Hsu)于 2021 年正式成立了 Arc Institute。

Patrick Collison 于 2019 年 6 月宣布与 Silvana Konermann 订婚
在成立之初,Arc 就筹集到了 6.5 亿美元的投资,其中 5 亿美元来自 Collison,这个「亿万富翁出钱让科学家妻子不再为科研经费发愁」的举动当年便在该领域内引起广泛讨论。这笔资金要为 15 名核心研究人员提供长达 8 年的资助,以及一个研究助理团队。这些研究者不受限制,可以以任何形式开展人类复杂疾病的研究。
这个专注于生命科学前沿研究和创新的非营利性科研机构命名源自 Island arcs(岛弧)。岛弧是板块与板块交界处隆起形成的群岛,创始人希望通过岛弧研究所集合许多不同机构、不同学科的研究人员,去创造一些新的东西。事实也的确如此,自成立以来,Arc Institute 面向生命科学领域推出了一系列重磅成果。
今年 2 月, Arc 研究所发布 Arc 虚拟细胞图谱,初始整合超 3 亿个细胞数据。该图谱首次推出两个基础数据集,于 2025 年 2 月 25 日开源开放:Tahoe – 100M 是由 Tahoe 创建的全新开源扰动数据集,含 1 亿个细胞及 50 种癌细胞系中的 6 万种药物 – 细胞相互作用;scBaseCount 是首个来自公共数据的单细胞 RNA 测序数据集,Arc 通过 AI 代理从公共存储库挖掘处理了代表 21 个物种的 2 亿多个细胞观测数据并标准化处理。
同年 4 月,10x Genomics、Ultima Genomics 与 Arc 研究所合作,加速开发 Arc 虚拟细胞图谱,其可计算的单细胞测量数据集合正通过 10x 和 Ultima 的技术不断增强。通过借助 10x 的 Chromium Flex 技术,以最低单细胞成本和最高分辨率大规模生成扰动数据,助力构建生物学 AI 模型;利用 Ultima 的 UG 100 测序系统及 Solaris 化学技术,以更低成本生成更多数据,还将使用 UG 100 Solaris Boost(一种目前处于早期访问阶段的全新高通量操作模式),以进一步提高数据产出。
向前追溯,2024 年 11 月,Arc 研究所联合斯坦福大学与加州大学伯克利分校开发出 Evo,这是首个基于 DNA 大规模训练的生物基础模型。它借助深度学习架构解析 DNA 编码信息,能在 DNA、RNA 及蛋白质层面进行预测与设计,覆盖从核苷酸到基因组的生物尺度,其核心价值在于破译 DNA 进化模式。研究团队利用其设计出自然界未知的功能性 CRISPR 系统 EvoCas9-1,仅测试 11 个设计便获得成功,其序列与常用 Cas9 相似度 73% 却具相当活性;此外,还成功设计了可移动的遗传元件 IS200/IS605 转座子。被誉为生物学领域的生成式 AI 基础模型。
2025 年 2 月,Arc 研究所在此基础上与 NVIDIA 合作开发迄今为止最大的生物学 AI 模型——Evo 2。Evo 2 基于超 10 万个物种的 9.3 万亿个核苷酸训练,能够识别基因序列模式,准确预测人类致病突变,还可设计相当于细菌基因组长度的新基因组。在技术上借助 NVIDIA DGX Cloud 平台的 2,000 余块 H100 GPU 训练,采用 StripedHyena 2 架构,处理数据量较前身 Evo 1 提升 30 倍,可同时分析百万核苷酸序列。
此外,2024 年 7 月,Arc 的 Goodarzi 实验室与 Gilbert 实验室合作,发现 mRNA 可以利用新发现的「RNA 开关」主动控制自身表达。2024 年 6 月,Arc 的 Hsu 实验室发现了第一个天然的 RNA 引导重组酶,它可以可编程地插入、切除或反转任何两个感兴趣的 DNA 序列,这是首个利用非编码 RNA 进行序列特异性靶向和供体 DNA 分子筛选的 DNA 重组酶,由于这种桥接 RNA 是可编程的,所以允许用户指定任何所需的基因组靶序列和任何待插入的供体 DNA 分子。
参考资料:
1.https://arcinstitute.org/news
2.https://mp.weixin.qq.com/s/THQTl2HI0mAXXwyykkQI5w



戳“阅读原文”,免费获取海量数据集资源!
(文:HyperAI超神经)