


作者:裴钰
指导/审阅:龚经经 龙思宇 范楷靖
本文由清华 AIR GenSI 研究组出品,转载请联系本公众号获得授权
清华大学 AIR GenSI 研究组联合清华大学药学院共同提出了一款用于蛋白质家族特异性生成建模的工具——ProfileBFN(即轮廓贝叶斯流网络),相关成果入选了 ICLR Oral 2025。
清华大学 AIR GenSI 研究组联合清华大学药学院共同提出了一种用于蛋白质家族特异性生成建模的工具——ProfileBFN(即轮廓贝叶斯流网络)。ProfileBFN 能够从多序列比对 (MSA) 轮廓的角度扩展了离散贝叶斯流网络,实现了高效的蛋白质家族设计。实证结果表明,在生成多样且新颖的家族蛋白质时,ProfileBFN 能够准确捕捉家族的结构特征。


论文链接:
https://go.hyper.ai/Dg5ha
关注公众号,后台回复「蛋白质家族 」获取完整 PDF
开源项目「awesome-ai4s」汇集了 200 余篇 AI4S 论文解读,并提供海量数据集与工具:
https://github.com/hyperai/awesome-ai4s
多序列比对: 蛋白质结构预测的基石
多序列比对 (MSA) 指的是将 3 种或更多生物序列 (DNA、RNA 或蛋白质) 进行比对的过程。进行多序列比对有助于发现和识别由于功能、结构或进化关系而产生的相似区域,为生物大分子之间关系提供更加全面的视角。
近年来,利用 MSA 信息已经成为蛋白质设计中的重要一环。在 AlphaFold、ESM 等里程碑式工作中都有专门模块编码 MSA 信息:
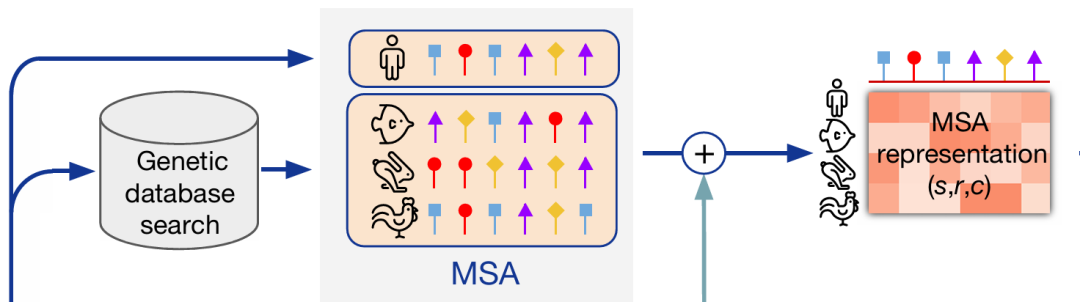
AF2 使用 MSA 作为一个提取特征的重要工具

MSA-Transformer in ESM
成也多序列,败也多序列
MSA 是进化信息的宝库,但是现有模型好像都高估了自己的挖宝能力。随着技术的发展,深度生成模型输入的 MSA 深度不断增加,但是效果却遇到了瓶颈,这让添加 MSA 信息的性价比遭到了质疑。其中根本原因在于 MSA 的数量和质量都存在严重的不确定性:
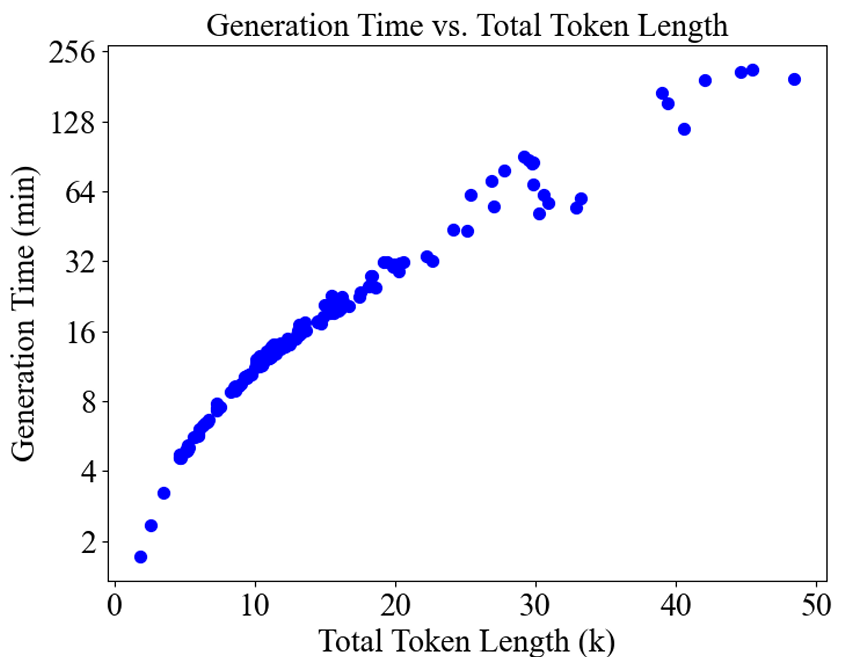
当 MSA 作为提示词输入到大模型中效率随输入长度的关系
研究人员将在多序列比对中相似于满足一定相似度的序列叫做同源序列。在数量上,对于某些「孤儿」蛋白质,同源序列可能不超过 10 条,而有些蛋白质能够搜索到超过 10,000 条同源序列,这给大模型造成了很大困惑,产生了资源的浪费和效率上的影响。
实际上,大自然的鬼斧神工岂是人类能妄加揣测的。在亿万年间的进化中,趋同结构反映了自然选择的效果,而变异则提供了进化新的可能性。对于这些特殊环境的特殊物种,它们往往保留了进化树伊始的原貌信息,这恰恰是共进化理论推演的基础。把同源序列作为模型输入的话,这些信息注定被大量其他无关信息所淹没,只能建模高概率的表示。为解决这一点,ProfileBFN 把每一簇同源序列建模成与数量无关的统一表示。
好的同源序列应该蕴含尽可能多的同源信息。实验表明,在大多数情况下,用几条信息熵最大的同源序列能起到和用上百条同源序列一样的效果。某些同源序列间仅有几个氨基酸的差别,它们给模型带来了很多误导的冗余信息。
Profile:下一代蛋白质基座模型基石
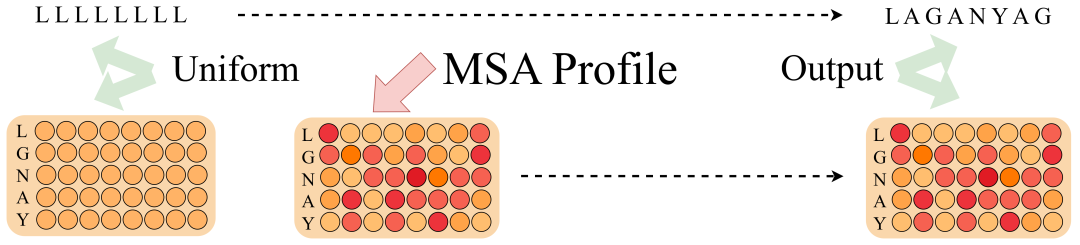
科学以发现为先,ProfileBFN 的创新的在于发掘原有 MSA 中存在的大量信息冗余。100 条同源序列,如果按照信息熵的方法排序,仅使用前 20 条训练就能让模型达到同样的效果。为此,一个单序列和多序列之间的桥梁需要被建立,这就是 Profile 出现的原因:
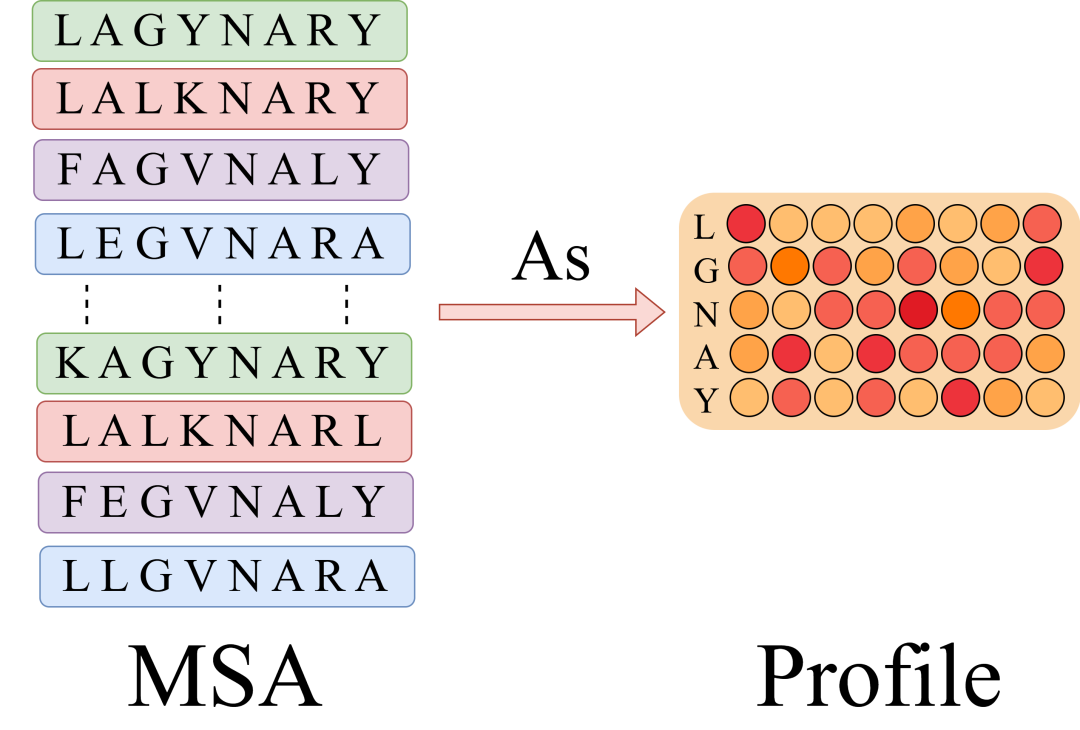
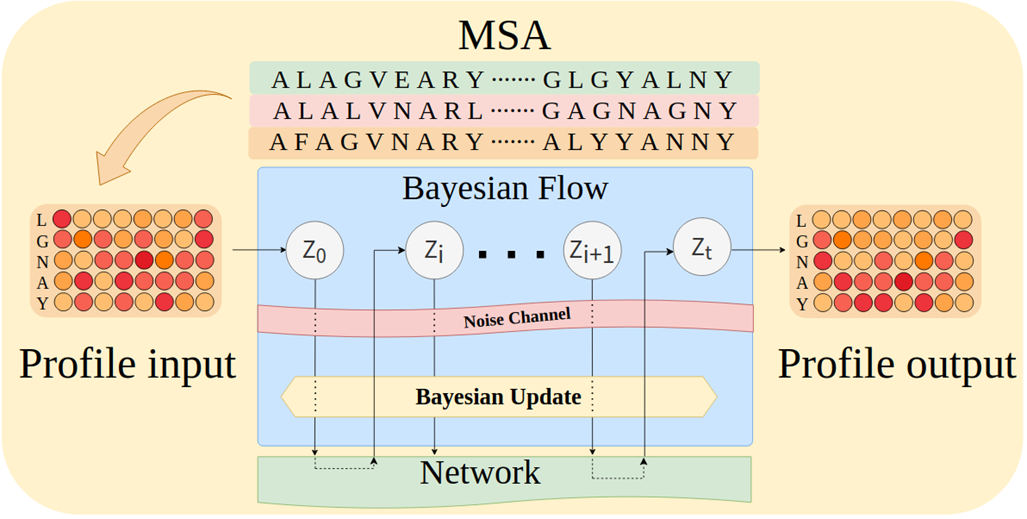
直观来理解,Profile 就是一个多序列比对中氨基酸出现次数的逐列统计。进一步说,如果有 1w 条同源序列,每条长度为 100,Profile 将其从 [10000,100] 直接压缩成了 [20,100] 的列表(20 种常见氨基酸),这大大简化了计算复杂度。特别的,单序列也可以看作特殊的 Profile,只不过每列只有一个 1。

ProfileBFN 发现,进行 MSA 到 Profile 的压缩不但没有原本预料的严重信息损失,还大大提升了模型性能。这一点可以理解为:在构建 Profile 的大浪淘沙中,每条同源序列都对这个位置出现氨基酸种类进行了投票表决,掩盖细微矛盾凸显整体趋势。
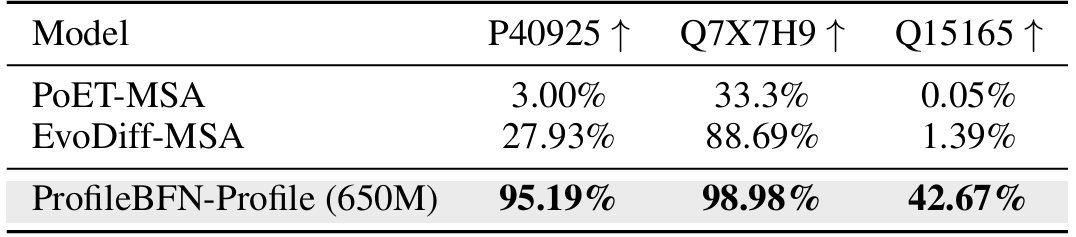
ProfileBFN 的强劲表现出人意料
相比于传统基于多序列比对的方法,ProfileBFN 依赖数据缩小 10 倍,学习到蛋白质序列上下文信息增加 1.5 倍,效果立竿见影!
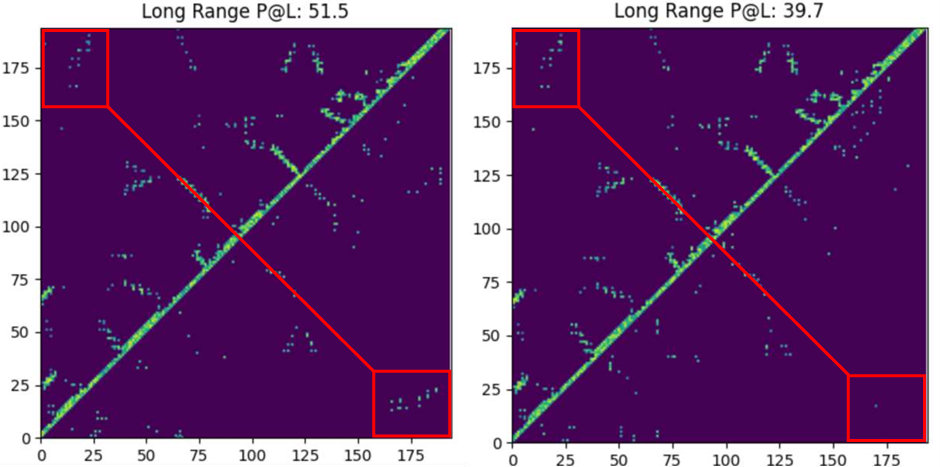
每幅图:左上是 GT,右下是模型预测上下文
左图: ProfileBFN 预测的上下文
右图:同源序列 (MSA) 预测的上下文
经过探索,已经证实 ProfileBFN 对多种下游任务都有促进作用:
* 酶分类:提升功能保真度,减少筛选成本
* 蛋白质表示学习:助力多任务特征提取
* 蛋白质结构预测:增强同源信息,提高建模精度
* 抗体生成:迁移作用优异,准确预测功能区域
酶是一类具有催化活性的特殊蛋白,其功能特异性通常通过 EC 编号 (Enzyme Commission Number) 进行描述。研究发现,ProfileBFN 生成的新酶候选在 EC 编号上高度匹配野生型酶,这意味着生成的蛋白质在功能上保持了高度一致性。这一特性大幅减少了实验筛选的难度,提高了新型酶设计的成功率。
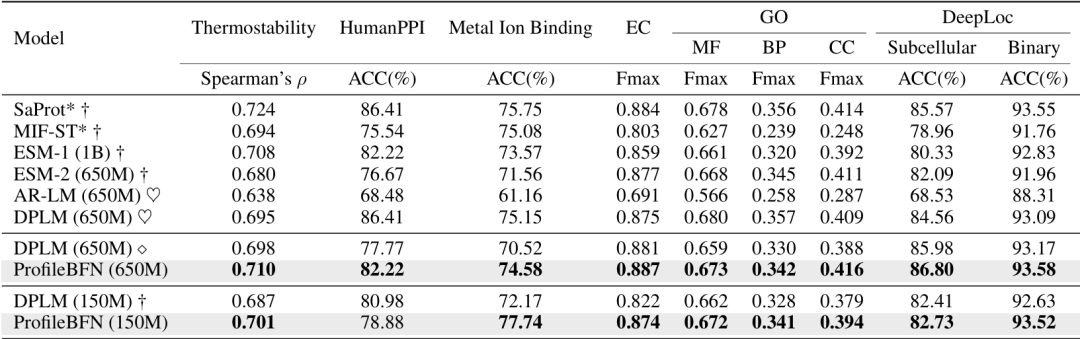
ProfileBFN 在生成蛋白质的同时,也在模型内部构建了精确的蛋白质表示。研究人员提取这些表示,并在蛋白质热稳定性、蛋白质相互作用、蛋白质亚细胞定位等多个数据集上进行微调,结果表明在分类等下游任务中,ProfileBFN 提供的表示能够有效提升模型性能。这表明它不仅是一种生成模型,还能作为强大的特征学习工具。
蛋白质结构预测是结构生物学的重要问题,尤其在孤儿蛋白质(即同源蛋白极少)场景下,传统方法的准确性受到极大限制。研究表明,ProfileBFN 可以作为同源信息增强器,在仅有少量 MSA 数据的情况下,生成更多高质量的同源蛋白质,从而提升 AlphaFold 系列模型的预测精度。这一能力使 ProfileBFN 在结构生物学领域展现出广阔的应用前景。
抗体是能与抗原特异性结合的功能性蛋白,在免疫和病理研究中具有重要意义。为探索 ProfileBFN 在抗体生成方面的潜力,研究人员基于 OAS (Observed Antibody Space) 抗体序列数据库 对模型进行了微调,结果显示 ProfileBFN 在生成多样化、高质量抗体序列方面表现出色。
ProfileBFN 的过人效果源于这一新研究给出了后 MSA 时代生成生物序列的范式:
* MSA 不直接作为输入参与训练过程,不引入额外训练开销
* 在推理阶段,对单序列和 MSA 进行统一建模
* 同源序列既是模型输入也是输出
BFN 完美利用先验信息
既然 Profile 信息很重要,甚至胜过原有同源序列,那么该如何利用 Profile 信息呢?贝叶斯流网络 BFN 对 Profile 的完美契合!这体现在两点:
* BFN 建模从分布到分布的过程,输入 Profile 表示输出仍然也是 Profile 表示
* 与其从零开始推理,BFN 可以引入 Profile 信息作为先验进行条件推理
对于自回归模型 (AutoRegressive)、扩撒模型 (Diffusion) 等传统模型,必须要求数据 (Tokens) 作为输入,处理 Profile 信息会额外增加算法复杂度。
有了 BFN 作为模型骨架,ProfileBFN 可以进而实现:
* 任务的简化。同源信息条件生成变为 Profile 信息模仿。
* 效率的提升。采样范围缩小有效性提高
ProfileBFN 有望成为湿实验救星
在合成生物学等任务中,周期长、评价指标单一、可信度不足是研究者们广泛遇到的问题。ProfileBFN 作为蛋白质基座模型,能够在资源有限的情况下整合更多同源信息,充分利用特定先验信息,对多指标有良好的迁移作用,这无疑让它成为合成候选蛋白,定向进化的不二之选。
关于研究组
清华大学智能产业研究院生成式符号智能研究组 (GenSI) 的研究领域横跨 LLM 和 AI for Science 两个方向,预期两个方向互相促进,从而实现 AGI for Science (AI Scientist) 的终极使命。
具体的研究方向包括新一代大规模预训练技术、超大规模强化学习 (Large Scale RL)、深度生成模型 (Deep Generative Models) 及其在科学数据中的应用等,注重人工智能基础算法和科学问题融合创新。目前,该团队聚焦于深度生成模型的前沿理论以及大规模结构化生成模型 (Scalable Structured-based Generative Models) 方法探索,致力于解决 LLM 和 AI4Sci 领域中现实且富有挑战的科学问题,例如提升 LLM 的推理能力、超越 AF3-level 的结构生成任务等。
点击查看团队解读深度生成模型发展脉络:「GenSI深度」|俯瞰深度生成模型发展脉络!从扩散模型,到流匹配再到贝叶斯流网络



戳“阅读原文”,免费获取海量数据集资源!
(文:HyperAI超神经)